


New Revelations around the Fraudulent Pfizer/BioNTech mRNA Vaccine Trial
A recent Pfizer data dump reveals more suspicious anomalies around the trial that launched a worldwide Covid-19 vaccination campaign

I have not written about the Covid-19 mRNA products recently. What’s the point?
The Pharma-owned medical establishment will likely never admit that mistakes were made. The CDC (Center for Disease Concoction and Perpetuation) continues to ignore the nearly quarter million reports of Serious Adverse Events (SAEs) and tens of thousands of reports of deaths following Covid-19 vaccination in their own adverse event reporting systems which were created as a concession to the public in exchange for granting vaccine manufacturers immunity from any liability related to their products.
Instead, the FDA and CDC have repeatedly doubled down on their assertion that these products, developed at a breakneck pace, have underwent the most rigorous testing for safety. How can anyone squeeze a typical five to ten year vaccine testing cycle into a few months? Is this why it was named Operation Warp Speed?
Nevertheless, by citing cherry-picked observational data riddled with confounders and flawed means of calculating effectiveness, the CDC and legacy media continue to assure the public that the mRNA vaccines are doing something good.
If there is ever going to be a reckoning it will come from an honest look at the trial that launched the worldwide vaccination campaign three years ago. Two recent papers analyze what has become recently available through a FOIA request submitted by PHMPT (Public Health and Medical Professionals for Transparency) in September, 2021.
Though their findings will likely not receive attention from legacy media, we can still endeavor to educate the public. This is why I have chosen to write this article.
The Trial was Suspicious from the Start
Much has been written about this multinational trial involving 44,000 participants over approximately six months. Though touted as a miracle of modern medicine at a time when we needed a miracle, formidable questions were asked and at least one whistleblower with evidence of fraudulent practices came forward. The questions went unanswered and the whistleblower was ignored.
How did we know that authorization of the Pfizer/BioNTech vaccine was unjustified three years ago? It came down to two pieces of data published in the NEJM paper that described the initial findings from the then on-going trial:
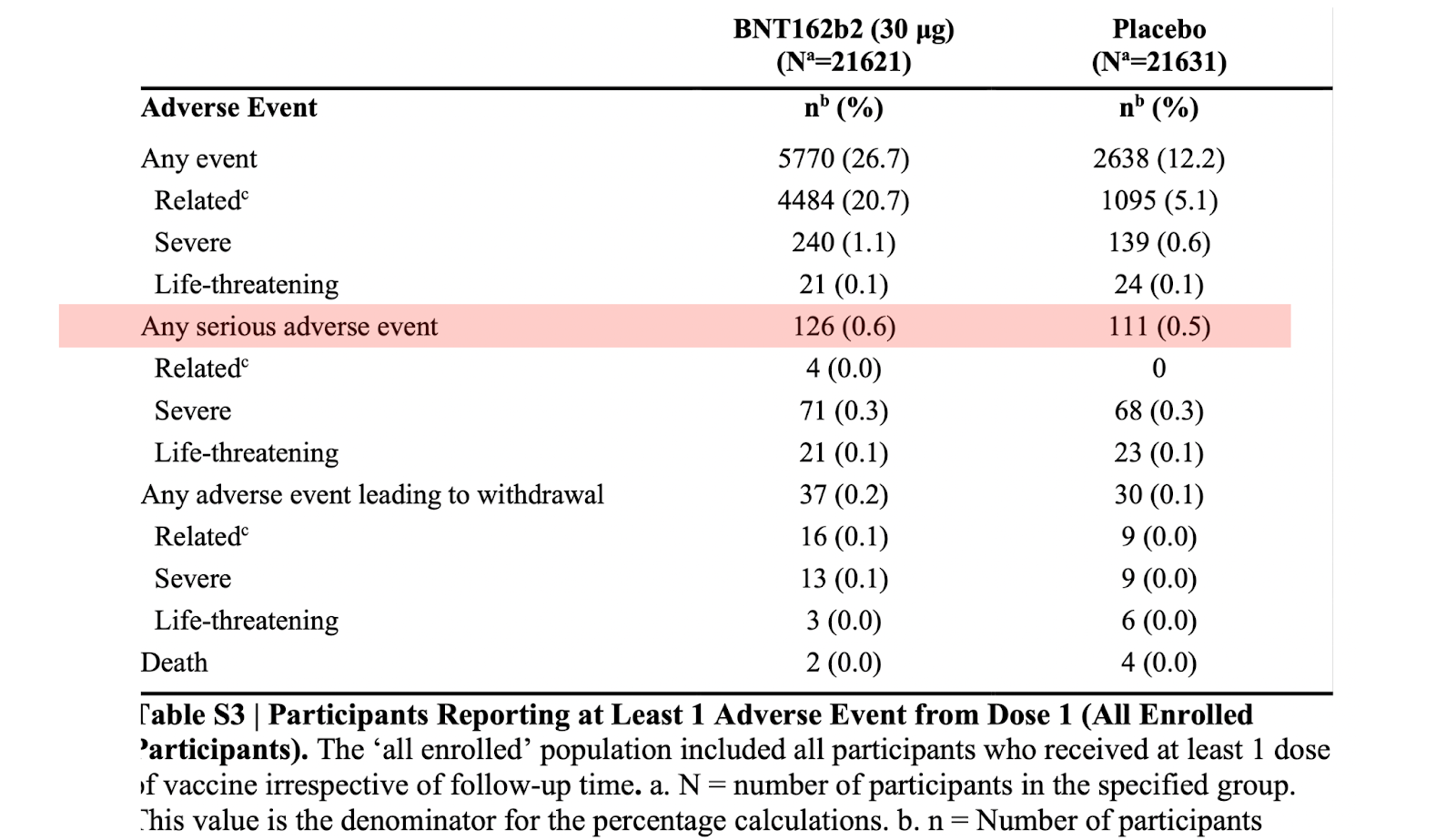
There were 126 out of approximately 22,000 vaccine recipients who suffered a Serious Adverse Event (SAE). Serious Events are, well, serious. Here is the definition:
SAEs are:
Death
A period where your life was threatened
The need for medical or surgical intervention
Hospitalization (initial or prolonged)
Permanent Disability
Birth Defect
126 out of 22,000 is 0.6%, or six in a thousand. Is this a high or low number? The answer is that it depends. It depends on the efficacy of the vaccine. Is it worth exposing yourself to that risk? Here is the answer:
From NEJM:
RESULTS
A total of 43,548 participants underwent randomization, of whom 43,448 received injections: 21,720 with BNT162b2 and 21,728 with placebo... Among 10 cases of severe Covid-19 with onset after the first dose, 9 occurred in placebo recipients and 1 in a BNT162b2 recipient.”
Injecting 21,720 people with the primary series will prevent eight cases of severe Covid-19 over the brief (six weeks) period of observation.
Severe Covid-19 is, well, severe. It puts you in the hospital. This is about the best we can do to compare apples to apples. The problem is that in order to prevent a single case of severe Covid approximately 2,500 people have to receive the primary series. That’s what the trial demonstrated.
This means that preventing a case of severe Covid-19 will result in fifteen SAEs! Another way to interpret this is that if you got the primary series there is a 1 in 2,500 chance that it will protect you from severe Covid if you expose yourself to a 1 in 167 (6 in 1000) chance of getting a SAE.
Why would anyone choose to take that bet? I understand that some would, for their own reasons, but why would the FDA and the CDC authorize a product with such a risk/benefit profile? Moreover, there was no reason to believe that the efficacy of the vaccine would improve over time. Like most vaccines, the mRNA shots were quickly shown to have waning efficacy. The number of SAEs, however, can only climb as time goes on. The odds that the primary series would do more good than harm was 15 to 1, and those odds would almost certainly get worse over longer periods of observation.
The answer is also in Table S3 (above). It turns out that 0.5% of those who got the placebo also had a SAE. That’s 5 in 1000. Because the difference in SAEs were relatively equal, advisory committees on the FDA and CDC concluded that the vaccine was relatively safe.
Pfizer explicitly states that the placebo used in their trial is saline, the safest compound to the human body, making it the ideal control to measure vaccine safety. As an anesthesiologist, I have personally given or attended to the administration of saline to over 20 thousand patients in my career. A SAE has never resulted.
The five in a thousand number is not representative of the risk of saline to human physiology; it represents the background rate of SAEs in the population, in this case, over a mean observational period of six weeks. Is this possible? I personally don’t think so, but this is difficult to prove.
We are not told which of the six types of SAEs made up the 111. We were also not told who suffered them. Were they healthy and young? Or old and sick? These are important factors when estimating the background rate of medical events. Most of the trial participants were healthy; only 1 in 5 had a previous medical condition.
There’s something screwy about that number of SAEs occurring in a pool of relatively disease free people in such a short time. We may now have some clues about why this happened…
Deeper Dives into the Trial Data
It’s been two years since PHMPT demanded that the FDA release the complete clinical trial data it relied upon when it licensed the Pfizer vaccine. A federal Court judge dismissed FDA objections and ordered the regulatory agency to comply, which it has been doing incrementally.
The most recent release of data centers around detailed narrative reports of participants who died or suffered adverse events during the trial.
Paper #1
Tore Gulbrandsen, a data scientist and guest author on Norman Fenton and Martin Neil’s substack, “Where are the Numbers” published this analysis of the recently released data:

He made a number of important observations. First was the fact that the total exposure measured in person years was 1.55 times higher in the vaccine arm. This means that if death rates in both arms of the trial were equal there would be 1.55 times more deaths among those who received the vaccine. There weren’t. Although the total number of deaths among the vaccinated were higher (21 vs 17), the mortality rate was lower in those who were vaccinated. This is indicative of a mortality benefit if a participant received the vaccine. This benefit was not statistically significant.
Next, he found a peculiar distribution of deaths in trial participants:

Figure 4 demonstrates that there is a statistically significant aberrancy in the timing of deaths from inoculation. There is a period of very few deaths soon after inoculation, but around 100 days later a large number of fatalities occur in a short window of time. How large is large? Plot C shows us the p-value for this anomaly. In other words, there is a less than 5% chance that this could have happened by coincidence alone.
This suggests that there may be a risk of vaccine mediated death that manifests after approximately 100 days. Gulbrandsen addresses the possibility that this might be due to a seasonal variation in death rate, however the correlation is stronger in elapsed time than in calendar time. The opposite would be true if this was a seasonal phenomenon.
More surprising is that this is happening in the placebo group at an even more improbable rate. There can only be three reasons for this. The placebo is responsible for these fatalities (impossible if the placebo is saline), the placebo recipients were reassigned (fraudulently) to the vaccine cohort (or both). Though highly improbable (1 in 500), there is always a chance that it happened coincidentally.
There is more data that suggests danger from the placebo:

Figure 6 is called a Forest plot. It depicts the ratio of the number of placebo recipients who suffered an adverse event to the number of vaccine recipients who did as well. As is demonstrated, nearly every category of participant medical history is overrepresented in the placebo group, including those with no previous medical conditions. This is reflected in the last row, “Sum”. There were 15.6% more placebo recipients who had an adverse event. This number is statistically significant.
Trials do not attribute causation, only correlation. The experimental vaccine is correlated with a lower risk of developing symptomatic and severe Covid-19. The same data indicates that the placebo is correlated with a higher incidence of adverse events. How is this possible?
Furthermore, Gulbrandsen found this:

Figure 5 plots the occurrence of adverse cardiac events in both groups. We see a peak of cardiac events in both groups 25 days after injection which is not statistically significant among placebo recipients. However, at approximately 100 days there is a statistically significant peak of cardiac events happening in the placebo group.
In other words, vaccinated participants had a peak of cardiac events three and a half weeks after injection. And, for some reason, placebo recipients had a surge of cardiac events and deaths approximately 100 days after injection which coincided with a surge of fatalities in the the vaccine group.
To summarize:
The placebo is associated with more adverse events than the mRNA vaccine
Fatalities and cardiac events are clustered around 100 days after injection in both cohorts
Though there were few fatalities in the days after injection in both groups, vaccine recipients had significantly more non-fatal cardiac events during this period
How are we to make sense of this?
We can only speculate.
If the placebo was cardiotoxic, there should have been a similar number of cardiac events in the placebo group soon after injection. There wasn’t.
If the placebo was innocuous, there shouldn’t have been a greater number of adverse events nor a large number of fatalities occurring at 100 days out in that group. There was.
One possibility is that the placebo was, in fact saline, but once injuries started occurring amongst the vaccinated, some were reassigned to the placebo group. This would explain the peak of mortality in both groups as well as the increased incidence of adverse events in the placebo group. What do you think (please comment)?
Paper #2
From a recently published article in the International Journal of Vaccine Theory, Practice and Research, “Forensic Analysis of the 38 Subject Deaths in the 6-Month Interim Report of the Pfizer/BioNTech BNT162b2 mRNA Vaccine Clinical Trial” (emphasis is mine):
This report focuses on the 38 trial subjects listed in the Pfizer/BioNTech 6-month Interim Report (6-Month Interim Report of Adverse Events C4591001) who died between the start of the trial on July 27, 2020 and March 13, 2021, the data end date of the 6-Month Interim Report.
Our analysis revealed important inconsistencies between the subject data listed in the 6-Month Interim Report and the materials on this data submitted by Pfizer/BioNTech to the FDA:Pfizer/BioNTech’s FDA Application for Emergency Use Authorization (Emergency Use Authorization for an Unapproved Product Review Memorandum Https://Archive.Org/Details/Emergency-Use-Authorization-Eua-for-an-Unapproved-Product-Review-Memorandum), Polack et al. (Polack 2020), and Thomas et al.(2021).
Most alarming, we found evidence of an over 3.7-fold increase in number of deaths due to cardiac events in BNT162b2 vaccinated subjects that Pfizer/BioNTech did not report. Had this information been known at critical time points, it might have been sufficient to question the safety of the BNT162b2 mRNA vaccine, delay EUA approval of the vaccine, and alter recommendations made to the public during the worldwide roll-out.
Here’s a key graph:

There are two big takeaways. The first is that throughout the course of the trial, the number of deaths in both arms of the trial nearly match each other. This is not what one would expect in a trial where one group was purportedly receiving a life-saving intervention and the other wasn’t.
The second is that the total number of deaths is actually well below what should have happened in a group of 44,000 people over this period of time, especially during the height of a pandemic. Based on age adjusted US death rates, the authors calculate that there should have been over 200 deaths in this period under normal circumstances.
The most likely explanation for this discrepancy is the large number of participants that were “discontinued subjects”. This group comprises 1 in 25 of the total number of people enrolled in the trial. Among that group are those who were “lost to follow up”. Approximately 100 in each group were lost to follow up prior to November 14, 2020, the cut-off date for data submitted by Pfizer to the FDA in support of their request for EUA.
There were only 11 deaths reported at that time. There should have been six times as many. With so few deaths reported, only a handful of deaths in either group of those who were “lost to follow-up” would have skewed a key metric in one way or another. Undoubtedly some of those who were lost to follow up died. How hard did Pfizer’s investigators investigate? Nevertheless, FDA regulators did not take issue with the missing participants.
Pfizer delayed death reports prior to Authorization
The Narrative Reports from individual trial sites demonstrate another suspicious pattern. The actual dates of death as reported in the Narrative Reports by investigators at each trial site do not match those of what Pfizer inserted in corresponding Case Reports. Pfizer used dates of death in the Case Reports to summarize the results of the ongoing trial to FDA regulators.
More importantly, the average delay in Case Reports was nearly three times higher among the vaccinated than the control group prior to authorization. But after EUA was granted, reporting delays dropped significantly in both groups. The end result was that two cardiac related deaths in the vaccinated group were omitted from consideration at the critical time of FDA EUA approval.
Furthermore there was a 25 day period prior to authorization when Pfizer, though aware of more fatalities, chose to not update the death count in the trial. Once again the FDA was not interested in the latest numbers. As a result six extra deaths were excluded from consideration at the time EUA was granted. Had they been included, a clearer picture of possible cardio toxicity would have emerged: 75% of deaths in the vaccine group were due to cardiac events compared to 33% of deaths in the placebo group. This was a clear safety signal that was hidden and would have prompted a more detailed inquiry assuming that the FDA was interested in doing their job.
Authors’ Conclusions
The authors summarize (emphasis is mine):
The C4591001 placebo-controlled randomized clinical trial of 22,030 vaccinated and 22,030 placebo subjects was the world’s only opportunity for an unbiased evaluation of the Pfizer/BioNTech BNT162b2 vaccine.
Unblinding of placebo subjects starting in Week 20 terminated the placebo-controlled clinical trial, thereby ending all unbiased evaluation of possible adverse event signals.
The mRNA-LNP platform is novel, not previously phase 2/3 tested in humans, and the toxicity of PP-Spike protein was unknown. Taken together, a 20-weeks placebo-controlled clinical trial is NOT sufficient to identify any except for the most common safety concerns.
The number of all-cause deaths is NOT decreased by BNT162b2 vaccination.
Of the 38 deaths reported in the 6-Month Interim Report of Adverse Events, 21 BNT162b2 vaccinated subjects died compared to 17 placebo subjects.
Delayed reporting of the subject deaths into the Case Report Form, which was in violation of the trial protocol, allowed the EUA to proceed unchallenged.
The number of subject deaths was 17% of the expected number, based on age-adjusted US mortality. One possible explanation could lie in the 395 subjects that were“Lost to Follow-up”.
There was a 3.7-fold increase in cardiac events in subjects who received the BNT162b2 vaccine versus the placebo.
Of the 15 subjects who were Sudden Adult Deaths (SAD) or Found Dead (FD), 12 died of a cardiac event, 9 of whom were vaccinated.
The cardiac adverse event signal was obscured by delays in reporting the accurate date of subject death that was known to Pfizer/BioNTech in the subject’s Narrative Report.
Conclusion
Three years later the results of the Pfizer trial are still hard to explain. With hundreds of thousands of pages of original data and narrative reports to examine, more clues may emerge with time. We can certainly say that the FDA and CDC advisory panels could not have adequately assessed this much information in the twenty five days they had prior to their decision to authorize this product for hundreds of millions of people.
Furthermore, a court order shouldn’t be necessary to impel the FDA to let the public know what can be known. What possible excuse would an agency of public health have for hiding data from the public?
These were not mistakes. This technology was given an unconditional green light from the highest levels.
If "mistakes were made", how did Moderna receive a contract on Jan 13, 2020, to produce an experimental "vaccine" designed over the weekend?
Why did SPARS and Event 201 dwell so extensively on countering "vaccine hesitancy" toward a vaccine that did not exist, for a disease class which had never been successfully vaccinated against?
The only mistakes here were made by the trusting public, who believed that their government would respond sensibly.
Interesting you brought up moderna. Right now all the negative news is strictly Pfizer with presence of sv40 & that they injected the population with an adulterated product.
Yet moderna did everything right?!
Apparently we’re supposed to be so distracted & excited that Pfizer may suffer & become extinct that we will not question moderna’s product.
Looks like a controlled demolition of Pfizer to take up all the bad consequences - injuries/deaths etc and then “they” can still hold up the MRNA platform as a viable vaccine product for humans & animals.